Welcome to genome-kmers’s documentation!
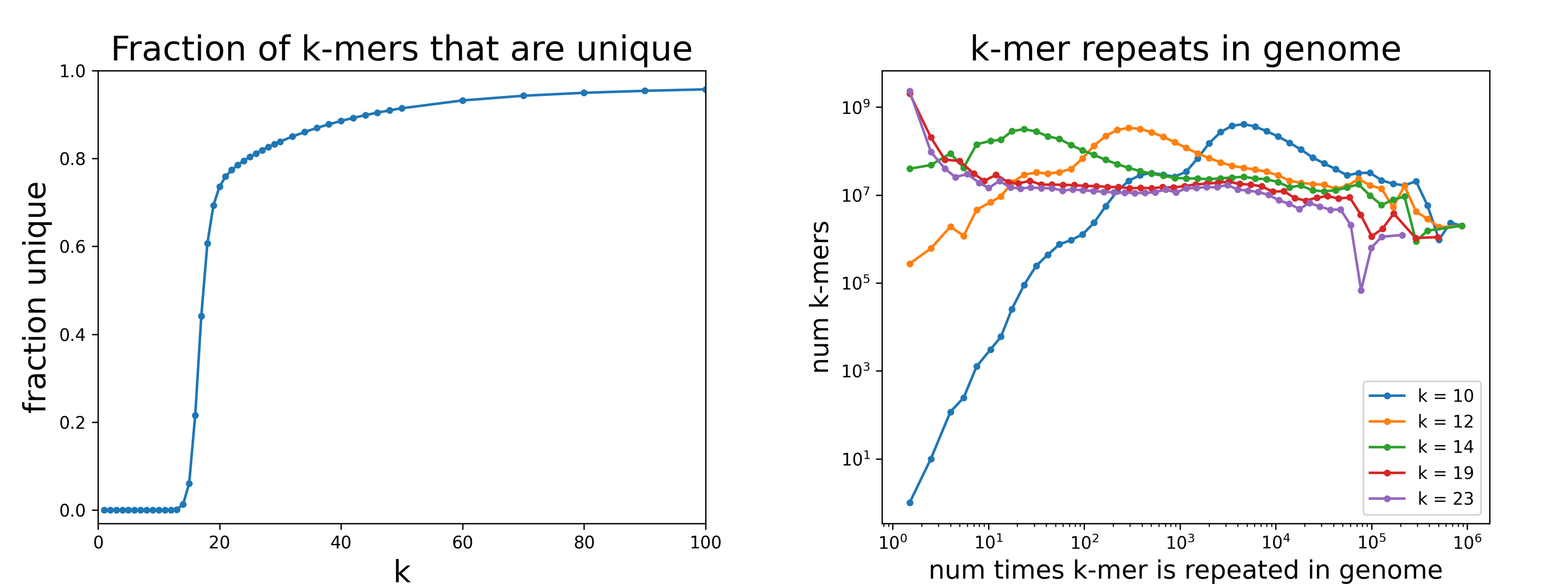
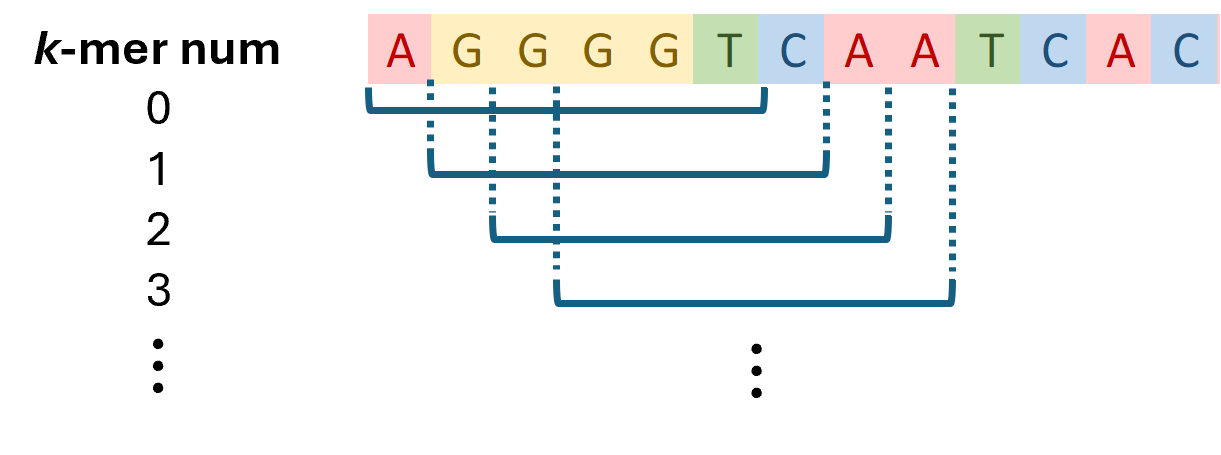
This package contains a collection of classes and methods useful for generating memory efficient k-mer statistics for a genome. Qualitatively, all the k-mers for a genome can be visualized as the sequence defined by a sliding window of width k.
Generating statistics is straightforward for smaller genomes (e.g. counting the number of unique 20-mers), but requires a different computational approach at the size of the human genome, which is around 3e9 base pairs. This package provides an efficient implementation with a simple user interface.